Osteogeneza imperfectă
Osteogeneza imperfectă este o afecțiune genetică. Este denumită în mod obișnuit boala oaselor fragile. Este o boală autozomal dominantă, ceea ce înseamnă că o persoană o poate contracta dacă doar unul dintre părinți are gena anormală. OI afectea…
Osteogeneza imperfectă este o afecțiune genetică. Este denumită în mod obișnuit boala oaselor fragile. Este o boală autozomal dominantă, ceea ce înseamnă că o persoană o poate contracta dacă doar unul dintre părinți are gena anormală. OI afectează partea din oase numită tija de colagen, care asigură rezistența oaselor. Gena anormală slăbește sau chiar distruge tija de colagen. Această boală a fost identificată pentru prima dată de Vrolik în 1854.
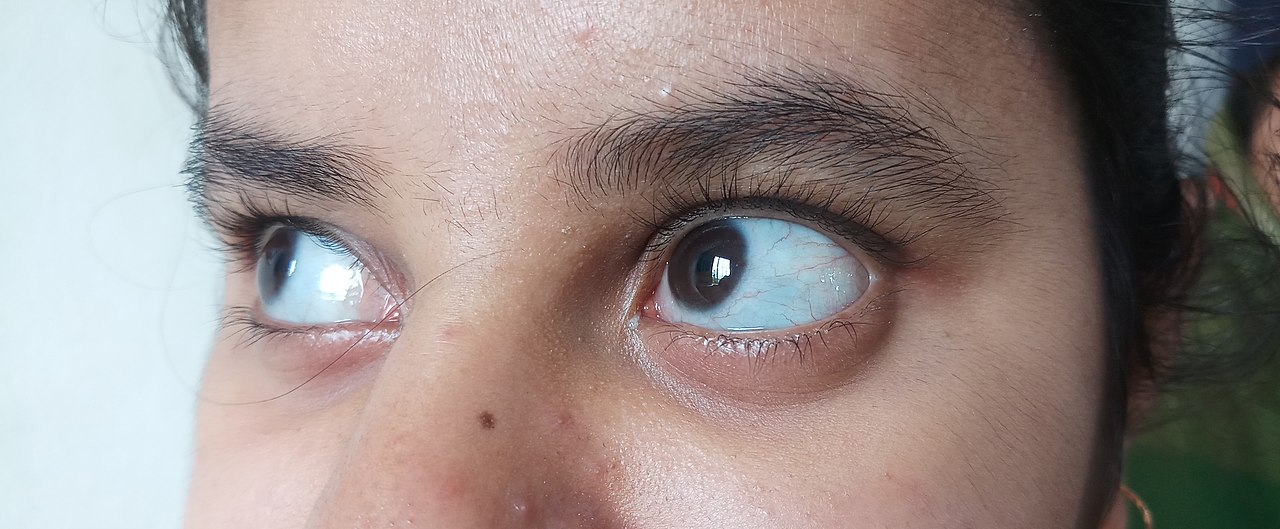
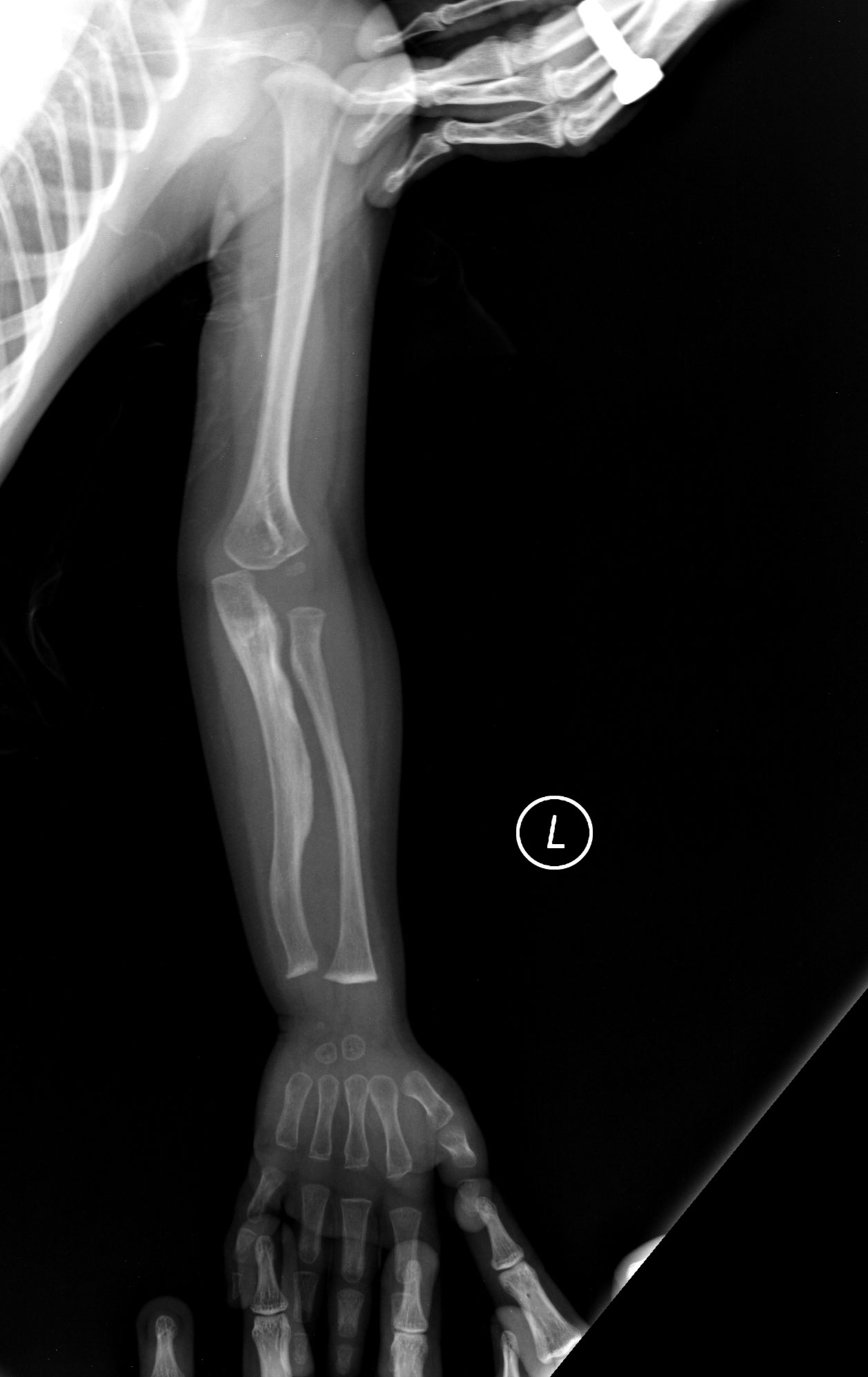
Osteogeneza imperfectă este o boală care apare atunci când cineva are oase slabe și este o afecțiune genetică. Atunci când cineva are această boală, este foarte probabil ca oasele să se rupă. Din păcate, Osteogeneza Imperfectă nu este vindecabilă. Această boală este clasificată în patru tipuri: Tipul unu este cel mai frecvent caz și cauzează adesea dinți casanți și leziuni legate de oase. Tipul doi este o versiune mai severă a tipului unu și prezintă majoritatea acelorași simptome, doar că în cazuri mai grave. Persoanele cu tipul trei de Osteogeneză Imperfectă pot avea mai mult de 100 de fracturi înainte de pubertate. Ochii lor dezvoltă adesea o nuanță purpurie, albastră sau gri, iar persoanele cu acest caz au, de asemenea, adesea pierderea auzului. 50% dintre persoanele care au Osteogeneză Imperfectă au pierderea auzului în timp ce devin adulți. Tipul 4 este un caz mai sever al majorității simptomelor menționate mai sus.
Galerie de imagini
10 Imagini







Simptome
Simptomele mai puțin severe ale OI pot include:
- oase ușor de rupt
- articulații slăbite
- tonus muscular scăzut
- culoarea albastră, violet sau gri a părții albe a ochilor, care în mod normal este albă
- forma triunghiulară a feței
- tendința de a dezvolta scolioză
- dinți fragili
OI are multe alte simptome grave și fatale, inclusiv probleme respiratorii și deformări osoase.
Date demografice
OI apare în egală măsură atât la bărbați, cât și la femei și poate afecta toate grupurile etnice. OI apare în uter și nu există tratament. De obicei, este descoperită atunci când cineva își rupe multe oase; este posibil să i se facă un test ADN pentru a verifica dacă are OI. Apare la 1 din 20.000 de nașteri.
Întrebări și răspunsuri
Î: Ce este osteogeneza imperfectă?
R: Osteogeneza imperfectă este o tulburare genetică numită în mod obișnuit boala oaselor fragile. Aceasta slăbește sau distruge tija de colagen, care asigură rezistența osoasă și duce la oase care sunt mai predispuse să se rupă.
Î: Cum se moștenește osteogeneza imperfectă?
R: Osteogeneza imperfectă este, de obicei, o boală autosomal dominantă, ceea ce înseamnă că o persoană o poate contracta dacă doar unul dintre părinți are gena anormală.
Î: Cine a identificat prima dată osteogeneza imperfectă?
R: Vrolik a identificat prima dată osteogeneza imperfectă în 1849.
Î: Există un tratament pentru osteogeneza imperfectă?
R: Din păcate, osteogeneza imperfectă nu are leac.
Î: Care sunt cele patru tipuri de osteogeneză imperfectă?
R: Cele patru tipuri de osteogeneză imperfectă sunt tipul unu, tipul doi, tipul trei și tipul patru.
Î: Care sunt simptomele osteogenezei imperfecte de tip trei?
R: Persoanele cu tipul trei de osteogeneză imperfectă pot avea mai mult de 100 de fracturi înainte de pubertate. Ochii lor dezvoltă adesea o nuanță purpurie, albastră sau cenușie, iar persoanele cu acest caz au, de asemenea, adesea pierderea auzului.
Î: Cât de frecventă este pierderea auzului la persoanele cu osteogeneză imperfectă?
R: 50% dintre persoanele care au osteogeneză imperfectă au pierderi de auz în timp ce devin adulți.
Articole similare
Autor
AlegsaOnline.com Osteogeneza imperfectă Leandro Alegsa
URL: https://ro.alegsaonline.com/art/73422
Surse
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"